Testing Genetic Engineering Detection with Spike-Ins
By Jeff Kaufman 🔸 @ 2024-10-22T17:20 (+15)
This is a linkpost to https://naobservatory.org/blog/testing-with-spike-ins
Thanks to our collaborators Marc Johnson and Clayton Rushford at the University of Missouri for their critical work on the laboratory experiments and sample preparation for this test, and to Evan Fields, Mike McLaren, and Simon Grimm for their feedback on drafts.
Summary
The NAO has been developing an early warning system for pandemics, focusing specifically on stealth pathogens that could spread widely before demonstrating identifiable symptoms. In June we described progress with our chimera detection approach, and demonstrated how it flagged engineered pathogens in simulated data and engineered viral vectors in real data. Here we describe a series of real-world tests, "spiking" samples of municipal wastewater with three different engineered HIV-derived viral particles. In each case our chimera detection system correctly identified sequencing reads containing the junctions between the original and modified sections of the genomes, with performance mildly exceeding our simulations.
Experiment
Our partners Marc Johnson and Clayton Rushford at the University of Missouri (MU) have been sequencing municipal wastewater influent on a regular basis. At the end of September they took one of these samples, split it into four replicates, and spiked HIV-derived engineered viral particles into three of them. The genomes of the viral particles looked something like the following:

The three particles were developed at MU, and each derives from the HIV-1 lab strain pNL4-3, with different inserted regions:
- v530: DsRed, a red fluorescent protein
- v549: Green Fluorescent Protein (GFP)
- Puro: a CMV promoter and a codon-optimized puromycin resistance gene
Our MU collaborators spiked 10uL into each of these samples. In the case of Puro the final concentration was 7.1k copies per mL. With v530 and v549, however, they haven't measured the concentration and so our assessment of those samples will be only in relative terms.
In addition to the three spiked samples they also prepared a control, a portion of the same original wastewater sample without the addition of any viral particles.
Our collaborators extracted the nucleic acids from these four samples using their standard protocol, which they describe in Defining biological and biophysical properties of SARS-CoV-2 genetic material in wastewater. They sent the extracts to the MU Genomics Technology Core for RNA sequencing on a NovaSeq X with the 10B flowcell.
Analysis
Once sequencing was complete we processed the data in several stages:
- We received several hundred million read pairs for each sample from the sequencing center as raw FASTQ files.
- We cleaned the data with AdapterRemoval2, removing adapters and collapsing overlapping forward and reverse pairs into merged reads.
- We used Bowtie2 to extract merged and unmerged pairs matching the ground truth genomes, and used Bowtie's alignments to deduplicate these read pairs. This filtered set we call the "Spike-in Pairs".
- To estimate how many reads an idealized chimera-detection pipeline could have flagged we further used Bowtie's alignment information to filter the Spike-in Pairs to ones with a forward, reverse, or merged read straddling one of the two junctions with at least 30bp of context on each side. We call these the "Junction Pairs".
- Separately, we ran the raw data through our chimera-detection pipeline, which flags read pairs that are only a partial match to known viral genomes. Of these, we call the ones matching the junctions in question "Flagged Pairs".
Table 1 shows the number of read pairs by sample at each stage as an absolute count, while Table 2 puts each stage in context by giving it as a fraction of the number of read pairs in the preceding column.
| Sample | Total Pairs | Spike-In Pairs | Junction Pairs | Flagged Pairs |
|---|---|---|---|---|
| v549 | 780M | 291,528 | 7,540 | 6,714 |
| Puro | 786M | 8,383 | 149 | 118 |
| v530 | 668M | 329 | 8 | 7 |
| Control | 840M | 3 | 0 | 0 |
Table 1: Number of read pairs by sample at each stage
| Sample | Total Pairs | Spike-In Pairs | Junction Pairs | Flagged Pairs |
|---|---|---|---|---|
| v549 | 780M | 0.04% | 2.59% | 89% |
| Puro | 786M | 0.001% | 1.78% | 79% |
| v530 | 668M | 0.00005% | 2.43% | 88% |
| Control | 840M | 0.0000004% | 0% | n/a |
Table 2: Fraction of read pairs at each stage as a fraction of the preceding column
While all four samples were sequenced to similar depths, the fraction of read pairs matching the spike-in genome in the three spike-in samples varied over multiple orders of magnitude. This is unsurprising because, as discussed above, of the three spike-in stocks only Puro was of a known concentration. The three Spike-In Reads in the Control sample are likely cross-sample contamination.
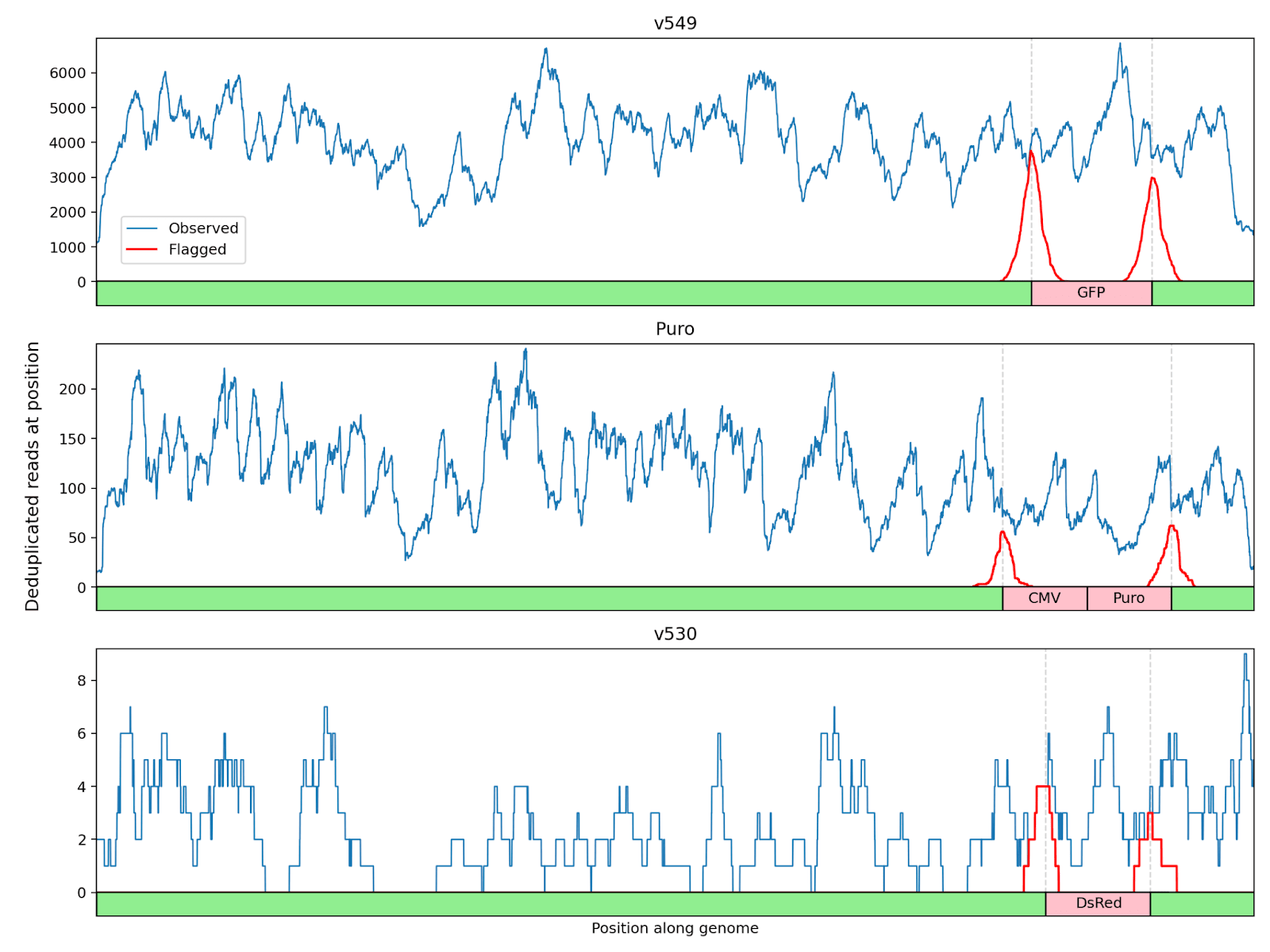
For each spiked sample, the following charts display two key pieces of information at each position along the spiked genome:
- The number of observed unique reads that cover that specific position with at least 30bp of context on each side.
- Of those reads, the number our chimera detection algorithm flagged as spanning one of the two suspicious junctions in that genome.
These charts show that our system flagged most, but not all, of the reads at each chimeric junction:
Discussion
In simulation on a range of viruses, our algorithm flagged an average of 1.2% of all reads from the pathogen genome and 71% of all reads that included the junction in context. Our results here are in the same range, but slightly better: an average of 2.2% of pathogen reads, and 85% of reads that included the junction in context. The improvement in the fraction matching the genome (1.2% → 2.2%) is primarily due to genome length, where the median genome we used in simulation was 15 kilobases and these three genomes are 8-9 kilobases: the shorter the genome is, the larger a fraction of reads will happen to include any individual position, and so more reads will cover each junction. The improvement in the fraction of flaggable reads that we did in fact flag (71% → 85%) is of course welcome, but we haven't yet dug into what is driving it.
Overall we see this experiment as quite positive: the system worked successfully end-to-end in flagging all three genetically modified viral particles that had been introduced into municipal influent, and it performed similarly to what our models predicted. With the very high metagenomic complexity of wastewater, this is an accomplishment we're proud of!